Computational Biology and Bioinformatics (CBB) Facility@CSIR-CDRI provides an integrated environment for informatics systems, computational chemistry and molecular modeling to facilitate and enhance drug design and discovery programs at CSIR-CDRI. CBB group’s research effort are focused on studying the macromolecules and their interactions with small molecules through the concepts and methods of the computational sciences. Computer-based methods in bio-molecular sciences are an increasing complement to experiment. Particular uses are for studying molecular details that are not easily accessible to experiment and making experimentally-testable predictions, especially for drug design.
We use structural bioinformatics and computational chemistry tools with specific focus on bio-molecular modeling and simulation, 3D-QSAR studies, virtual screening and development of computational predictive models towards the identification and design of small molecules as inhibitors/vaccines or chemical probes against various therapeutically important drug targets.
Capabilities/Focus Area of Research
➤Informatics: Bio-informatics, Chem-informatics and Phamaco-informatics.
➤ Evolutionary Bioinformatics: Phylogenetic Analysis etc
➤Computational Structural Biology: Biomolecular engineering & Modeling, Computer-Aided Molecular Design and Simulation, Quantum Chemical Calculations.
➤ Rational Drug Design: Virtual screening, de novo design, pharmacophore perception, SAR and ADME, Molecular docking, Conformational Analysis, Binding free energy simulations, Protein Ligand interactions etc.
Computational Resources
Computing Environment
HP Proliant DL385 Gen 8 Servers, SGI Graphics octane graphics workstations, SGI Origin 300 (4x32-bit processors) and Itanium- based SGI- Altix 350 Linux computer server (6x64 bit processors), Intel core-i3, i5,Xeon-based high performance workstations etc.
Software packages
Software packages including Accelrys (Discovery Studio, InsightII & Cerius2), Tripos (SYBYL & UNITY), GAUSSIAN and other computational tools for computer-aided drug design, bio-molecular modeling & simulation, management and mining of genetic, biological, chemical data and information.
Data Storage
SGI Total performance 9100 (TP100) high performance fiber channel storage array, Lenovo ix2-dl network storage.
Recent Significant Accomplishments
➤ Identification of Novel Inhibitors of Mycobacterium tuberculosis PknG Using Pharmacophore Based Virtual Screening, Docking, Molecular Dynamics Simulation, and their Biological Evaluation.
 The essential role of PknG in the pathogenesis and survival of the tubercle bacillus within host by preventing phagosome-lysosome fusion as well as in developing intrinsic antibiotic resistancemakes it an attractive drug target. Therefore, we carried out pharmacophore-based virtual screening and the proposed hits were subjected to in vitro experiments. Three out of six tested molecules showed significant inhibitory activity against MtPknG. In addition, inhibitory studies of mycobacterial growth in infected THP-1 macrophages demonstrated considerable growth inhibition of M. bovis BCG induced through compound NRB04248 without any cytotoxic effect against host macrophages, suggesting its suitability for further design and optimization of MtPknG inhibitors.
The essential role of PknG in the pathogenesis and survival of the tubercle bacillus within host by preventing phagosome-lysosome fusion as well as in developing intrinsic antibiotic resistancemakes it an attractive drug target. Therefore, we carried out pharmacophore-based virtual screening and the proposed hits were subjected to in vitro experiments. Three out of six tested molecules showed significant inhibitory activity against MtPknG. In addition, inhibitory studies of mycobacterial growth in infected THP-1 macrophages demonstrated considerable growth inhibition of M. bovis BCG induced through compound NRB04248 without any cytotoxic effect against host macrophages, suggesting its suitability for further design and optimization of MtPknG inhibitors.
Singh N, Tiwari S, Srivastava KK, Siddiqi MI., J Chem Inf Model. 2015 Jun 22;55(6):1120-9. doi: 10.1021/acs.jcim.5b00150
➤ Identification of novel inhibitors of human Chk1 using pharmacophore-based virtual screening and their evaluation as potential anti-cancer agents.
Kinases are one of the major players in cancer development and progression. Chk1 is an important kinase with vital role in cell cycle arrest and many potent inhibitors targeted to Chk1 have been reported and few are currently in clinical trials. In this work, we have reported an integrated in silico approach to identify novel competitive Chk1 inhibitors. A 4-features 3D-pharmacophore model was built on the basis of known Chk1 inhibitors belonging to thienopyridine series and virtual screening of Maybridge database and subsequent docking analysis of hits were carried out to identify potential hChk1 inhibitors. Further, 5 compounds from the top ranking hits were subjected to in vitro evaluation as Chk1 inhibitor. One of the hits, SPB07479 showed significantly inhibited proliferation of cervical cancer cells as a single agent and chemosensitized glioma and pancreatic cancer cell lines to standard chemotherapy while sparing normal cells. Additionally SPB07479 did not show significant cytotoxity in normal cells. Above finding supports that SPB07479 can be further explored to design and develop more potent Chk1 inhibitors.
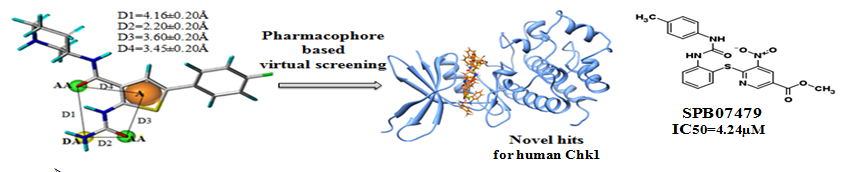
Kumar V, Khan S, Gupta P, Rastogi N, Mishra DP, Ahmed S, Siddiqi MI., J Comput Aided Mol Des. 2014 Dec;28(12):1247-56. doi: 10.1007/s10822-014-9800-9.
➤ Pharmacophore-based screening and identification of novel human ligase I inhibitors with potential anticancer activity.
Human DNA ligases are enzymes that are indispensable for DNA replication and repair processes. In the quest to develop new anticancer agents, we have employed a pharmacophore-based virtual screening approach for searching novel human Ligase I (hLigI) inhibitors within commercially available Maybridge database containing more than 56000 compounds. On the basis of previously reported inhibitors of the targeted protein, pharmacophore model was generated. The identified hits were subjected to docking into the active site of hLigI. The shortlisted five hit molecules identified through the pharmacophore-based virtual screening were purchased and subjected to biological assay. The selected compounds showed significant inhibition against hLigI. Our results demonstrate the efficiency of pharmacophore based virtual screening for the identification of potential hLigI inhibitors that also demonstrated potential anticancer activity against a colon cancer cell line.
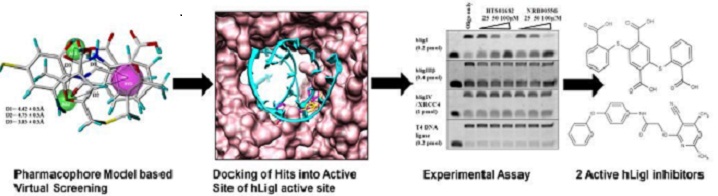
Krishna S, Singh DK, Meena S, Datta D, Siddiqi MI, Banerjee D., J Chem Inf Model. 2014 Mar 24;54(3):781-92. doi: 10.1021/ci5000032.
➤ Machine learning based web server for identification of potential anti-diabetic agents
To reduce the cost and time of drug discovery process, it is important to create some robust in-silico methods for rapid screening of large chemical libraries. In this context, we have built support vector machine (SVM) classification model to discriminate inhibitors from the non-inhibitors using available inhibitor and their activity information of known anti-diabetic targets. SVMDLF (Support Vector Machine based DPP4 Lead Finder) is a web server developed by our group for predicting Dipeptidyl peptidase 4 (DPP4) inhibitors based on SVM classification model.
Chandra, Jyotsna Pandey, Akhilesh K. Tamrakar, Mohammad Imran Siddiqi, SVMDLF: a web server for prediction of Dipeptidyl peptidase 4 inhibitors. (Manuscript to be communicated)
Availability: Link-I and Link-II
➤ Integrating molecular docking, CoMFA analysis and Machine Learning classification with virtual screening towards identification of novel scaffolds as Plasmodium falciparum enoyl acyl carrier protein reductase inhibitor.
An integrated application of various in silico methods including molecular docking, CoMFA analyses and machine learning classification methods were used on a set of known inhibitors of Plasmodium falciparum enoyl acyl carrier protein reductase (PfENR) and maybridge compound database with the prime objective of implementation of knowledge-based synergistic approach towards prioritized screening and identification of novel scaffolds as pfENR inhibitors. 26 compounds have been prioritized as potential anti-PfENR compounds on the basis of proposed workflow.
Shah, P., Tiwari, P. and Siddiqi, M.I., Medicinal Chem. Res. (2014), DOI: 10.1007/s00044-014-0910-7
➤ 3D-QSAR and molecular modeling studies on 2,3-dideoxy hexenopyranosid-4-uloses as anti-tubercular agents targeting alpha-mannosidase.
Ligand and structure-based methods were applied in combination to exploit the physicochemical properties of 2,3-dideoxy hex-2-enopyranosid-4-uloses for designing novel anti-tubercular agents. Furthermore, 3 selected inhibitors from this class of compounds with known anti-Mtb α-mannisidase activity were used for the molecular interaction analysis to correlate the proposed QSAR guidelines with molecular basis of binding affinity with the homology model on Zn-dependent Mycobacterium tuberculosis α-mannosidase. Synergistic complementation between the results obtained from both the approaches could be helpful in rationalizing and optimizing the anti-tubercular activities and design of new 2,3-dideoxy hex-2-enopyranosid-4-uloses analogs.
Shah P, Saquib M, Sharma S, Husain I, Sharma SK, Singh V, Srivastava R, Shaw AK, Siddiqi MI. Bioorg Chem. 2015 Apr;59:91-6. doi: 10.1016/j.bioorg.2015.02.001.
➤ Homology modeling of NAD+-dependent DNA ligase of the Wolbachia endosymbiont of Brugia malayi and its drug target potential using dispiro-cycloalkanones.
The antifilarial drug target potential of NAD+ dependent DNA ligase of Wolbachia (wBm-LigA) was investigated by docking studies of dispiro-cycloalkanone compounds in the homology modelled structure of NAD+ dependent DNA ligase of Wolbachia (wBm-LigA). Further effects of these inhibitors were observed on adult and microfilarial stages of B. malayi in vitro and the most active compounds were further followed in vivo in jirds (primary screen) and mastomys (secondary screen).
Shrivastava N, Nag JK, Pandey J, Tripathi RP, Shah P, Siddiqi MI, Misra-Bhattacharya S. Antimicrob Agents Chemother. 2015 Jul;59(7):3736-47. doi: 10.1128/AAC.03449-14
➤ Molecular Modeling studies on 3-Arylcoumarin-tetracyclic Tacrine Hybrids as Multifunctional Agents against Parkinson's Disease (PD)
 A series of 3-arylcoumarin-tetracyclic tacrine derivatives was designed and synthesized as potential leads against aging and age associated PD. A number of derivatives demonstrated significant reduction of aggregation of "human" alpha-synuclein (α-synuclein) protein, expressing on transgenic Caenorhabditis elegans (C. elegans) model NL5901. Our molecular modeling studies, complementing the experimental results predicted interaction of most active compounds of the series with α-synuclein protein.
A series of 3-arylcoumarin-tetracyclic tacrine derivatives was designed and synthesized as potential leads against aging and age associated PD. A number of derivatives demonstrated significant reduction of aggregation of "human" alpha-synuclein (α-synuclein) protein, expressing on transgenic Caenorhabditis elegans (C. elegans) model NL5901. Our molecular modeling studies, complementing the experimental results predicted interaction of most active compounds of the series with α-synuclein protein.
Koneni, S., Ram, M., Pooja, J., Rao, K.,Sharma, T., Haq, R., Singh, D., Banerjee, D., Siddiqi,M.I., Nazir, A.; ACS Med. Chem. Lett., 2014, 5 (10), pp 1099–1103 DOI: 10.1021/ml500222g
Recent Publications from the group
Please click the below links to see the complete list of Publications.
➤ Link-I
➤ Link-II
➤ Link-III
Opportunities for National /International collaborations with Institutions/Industries etc
CBB Facility @CSIR-CDRI is actively involved in research collaborations. Potential collaborations in the focus areas of research can be explored with CBB Facility@CSIR-CDRI.
Opportunities for Students
There are plenty of opportunities for bio-informatics/chem-informatics research projects for students at CBB facility@CSIR-CDRI. Various positions of JRF/Project Fellow/Project Trainee in the projects at the facility are advertised at CSIR-CDRI website from time to time and prospective students may contact the resource person accordingly.
Present Group Members
- ➤ Vikash Kumar (DBT-SRF)
- ➤ Nidhi Singh (CSIR-SRF)
- ➤ Sharat Chandra (UGC-SRF)
- ➤ Tanuj Sharma (CSIR-SRF)
|
- ➤ Shagun Krishna (ICMR-SRF)
- ➤ Kamakshi Sikka (UGC-JRF)
- ➤ Jitender Kuldeep (UGC-JFR)
- ➤ Sabahuddin Ahmed (Project Fellow)
|
Contact Resource Person
Dr. Mohammad Imran Siddiqi
Principal Scientist
Computational Biology & Bioinformatics Facility
LSN-104, Biological Sciences, North Block
Molecular and Structural Biology Division
CSIR-Central Drug Research Institute,
Lucknow- 226031, U.P., India
Email: mi_siddiqi@cdri.res.in