Significant Research and Findings:
A) Role of Interleukin-1 Receptor Associated Kinase (IRAK) in Monocyte/Macrophage Non-Sterile Inflammation
i) The IRAK-ERK-p67phox-Nox-2 axis mediates TLR4, 2-induced ROS production for IL-1β transcription and processing in monocytes.
In monocytic cells, Toll-like receptor 4 (TLR4)- and TLR2-induced reactive oxygen species (ROS) cause oxidative stress and inflammatory response; however, the mechanism is not well understood. The present study investigated the role of interleukin-1 receptor-associated kinase (IRAK), extracellular signal-regulated kinase (ERK), p67phox and Nox-2 in TLR4- and TLR2-induced ROS generation during interleukin-1 beta (IL-1β) transcription, processing, and secretion. An IRAK1/4 inhibitor, U0126, PD98059, an NADPH oxidase inhibitor (diphenyleneiodonium (DPI)), and a free radical scavenger (N-acetyl cysteine (NAC))-attenuated TLR4 (lipopolysaccharide (LPS))- and TLR2 (Pam3csk4)-induced ROS generation and IL-1β production in THP-1 and primary human monocytes. An IRAK1/4 inhibitor and siRNA-attenuated LPS- and Pam3csk4-induced ERK-IRAK1 association and ERK phosphorylation and activity. LPS and Pam3csk4 also induced IRAK1/4-, ERK- and ROS-dependent activation of activator protein-1 (AP-1), IL-1β transcription, and IL-1β processing because significant inhibition in AP-1 activity, IL-1β transcription, Pro- and mature IL-β expression, and caspase-1 activity was observed with PD98059, U0126, DPI, NAC, an IRAK1/4 inhibitor, tanshinone IIa, and IRAK1 siRNA treatment. IRAK-dependent ERK-p67phox interaction, p67phox translocation, and p67phox-Nox-2 interaction were observed. Nox-2 siRNA significantly reduced secreted IL-1β, IL-1β transcript, pro- and mature IL-1β expression, and caspase-1 activity indicating a role for Nox-2 in LPS- and Pam3csk4-induced IL-1β production, transcription, and processing.
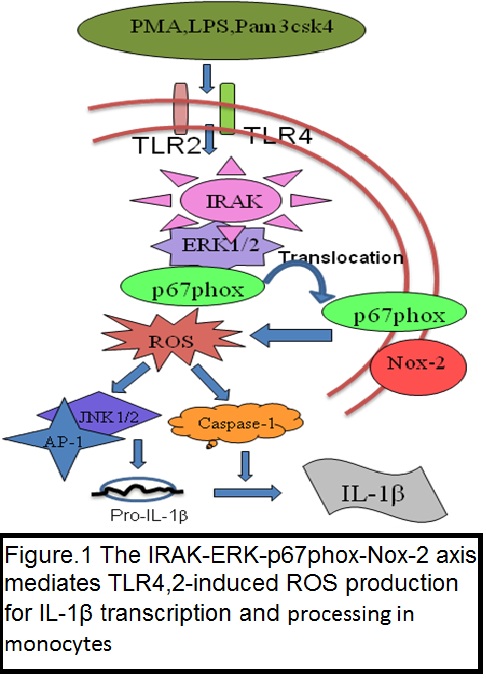
In the present study, we demonstrate that the TLR4- and TLR2-induced IRAK-ERK pathway cross-talks with p67phox-Nox-2 for ROS generation, thus regulating IL-1β transcription and processing in monocytic cells (Cell Mol Immunol. 2016 Nov;13(6):745-763).
B) Role of Interlukin-1-Receptor Associated Kinase (IRAK) and Oxidized Low Density Lipoprotein (Ox-LDL) in Sterile Inflammation, Macrophage Foam cell formation and Atherosclerosis:
i) IRAK Mediates Ox-LDL induced Sterile Inflammation:
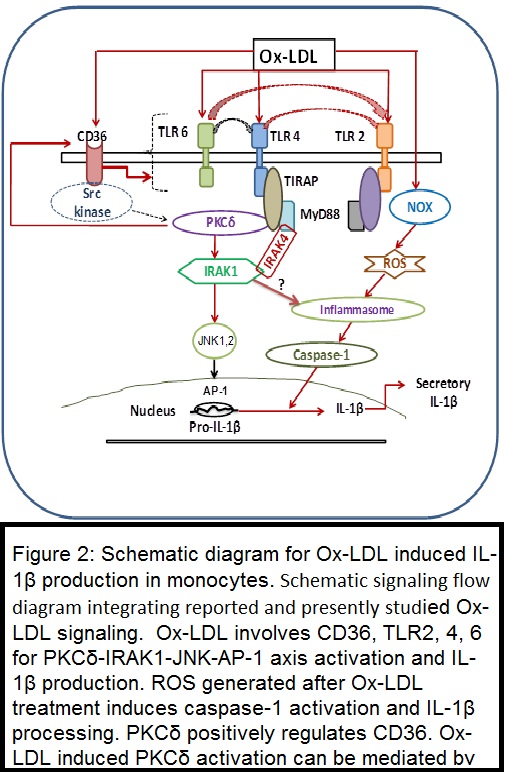
This study examined the role of interleukin (IL)-1 receptor-associated kinase (IRAK) and protein kinase C (PKC) in oxidized LDL (Ox-LDL)-induced monocyte IL-1β production. In THP1 cells, Ox-LDL induced time-dependent secretory IL-1β and IRAK1 activity; IRAK4, IRAK3, and CD36 protein expression; PKCδ-JNK1 phosphorylation; and AP-1 activation. In addition to this PMA treatment to THP1 cells induced CD11b, TLR2, TLR4, CD36, IRAK1, IRAK3, and IRAK4 expression, IRAK1 kinase activity, PKCδ and JNK phosphorylation, AP-1 and NF-κB activation, and secretory IL-1β production. IRAK1/4 siRNA and inhibitor (INH)-attenuated Ox-LDL and PMA induced secreted IL-1β and pro-IL-1β mRNA and pro-IL-1β and mature IL-1β protein expression, respectively. Diphenyleneiodonium chloride (NADPH oxidase INH) and N-acetylcysteine (free radical scavenger) attenuated Ox-LDL-induced reactive oxygen species generation, caspase-1 activity, and pro-IL-1β and mature IL-1β expression. In PKCδ wild-type overexpressing THP1 cells, IRAK1 kinase activity and IL-1β production were significantly augmented, whereas recombinant inactive PKCδ and PKCδ small interfering RNA significantly inhibited basal and PMA-induced IRAK1 activation and IL-1β production. Ox-LDL-induced secretory IL-1β production was abrogated in the presence of JNK INH II, Tanshinone IIa, Ro-31-8220, Go6976, Rottlerin, and PKCδ siRNA. PKCδ siRNA attenuated the Ox-LDL-induced increase in IRAK1 kinase activity, JNK1 phosphorylation, and AP-1 activation. In THP1 macrophages, CD36, toll-like receptor (TLR)2, TLR4, TLR6, and PKCδ siRNA prevented Ox-LDL-induced PKCδ and IRAK1 activation and IL-1β production.
Enhanced Ox-LDL and IL-1β in systemic inflammatory response syndrome (SIRS) patient plasma demonstrated positive correlation with each other and with disease severity scores. Ox-LDL-containing plasma induced PKCδ and IRAK1 phosphorylation and IL-1β production in a CD36-, TLR2-, TLR4-, and TLR6-dependent manner in primary human monocytes.
Results suggest involvement of CD36, TLR2, TLR4, TLR6, and the PKCδ-IRAK1-JNK1-AP-1 axis in Ox-LDL-induced IL-1β production (J Lipid Res. 2014 Jul;55(7):1226-44 and J Immunol. 187:2632,2011 )
ii) IRAK regulates macrophage foam cell formation by modulating genes involved in cholesterol uptake and efflux.
Interleukin-1 receptor-associated kinase-1 (IRAK1) is linked to the pathogenesis of atherosclerosis; however, its role in macrophage foam cell formation is not known. Therefore, the present study investigated the role of IRAK1 in lipid uptake, biosynthesis, and efflux in THP-1 derived macrophages and human monocyte-derived macrophages (HMDMs). Ox-LDL (40 μg/mL, 15 minutes-48 hours) treatment induced time-dependent increase in IRAK1, IRAK4, and Stat1 activation in THP-1 derived macrophages. IRAK1/4 inhibitor (INH) or IRAK1 siRNA significantly attenuated cholesterol accumulation, DiI-Ox-LDL binding, and uptake while cholesterol efflux to apoAI and HDL was enhanced in THP-1 derived macrophages and HMDMs. Ox-LDL treatment significantly increased the mRNA expression of CD36, LOX-1, SR-A, ABCA1, ABCG1, Caveolin-1, CYP27A1 while that of SR-BI was decreased. IRAK1/4 inhibition or IRAK1 knockdown, however, attenuated Ox-LDL-induced CD36 expression; augmented ABCA1 and ABCG1 expression while expression of others was unaffected in THP-1 derived macrophages and HMDMs. Moreover, IRAK1/4 inhibition had no significant effect on genes involved in lipid biosynthesis. In IRAK1/4 INH pre-treated THP-1 derived macrophages Ox-LDL-induced Stat1 phosphorylation and its binding to CD36 promoter was significantly attenuated while LXRα expression and its binding to the ABCA1/ABCG1 locus, NFATc2 activation and its binding to ABCA1 locus was enhanced.
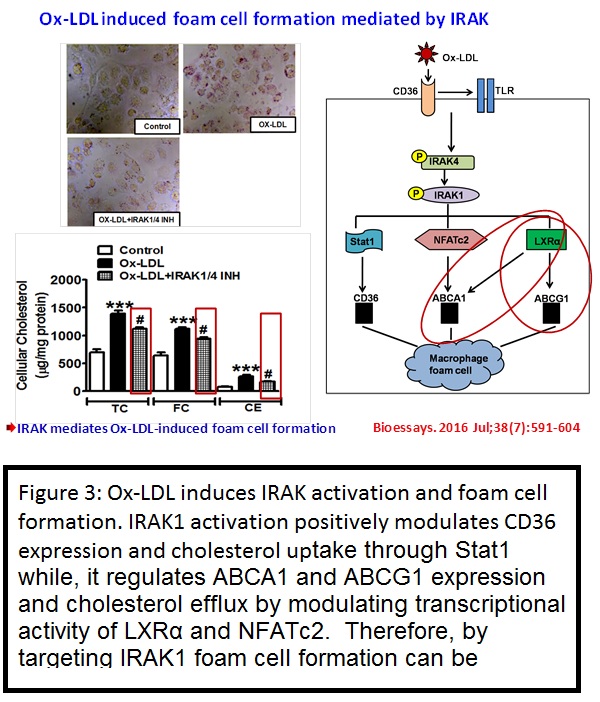
The present study thus demonstrates that IRAK regulates lipid accumulation by modulating CD36-mediated uptake and ABCA1-, ABCG1-dependent cholesterol efflux. Therefore, IRAK1 can be a potential target for preventing macrophage foam cell formation (Bioessays. 2016 Jul;38(7):591-604)
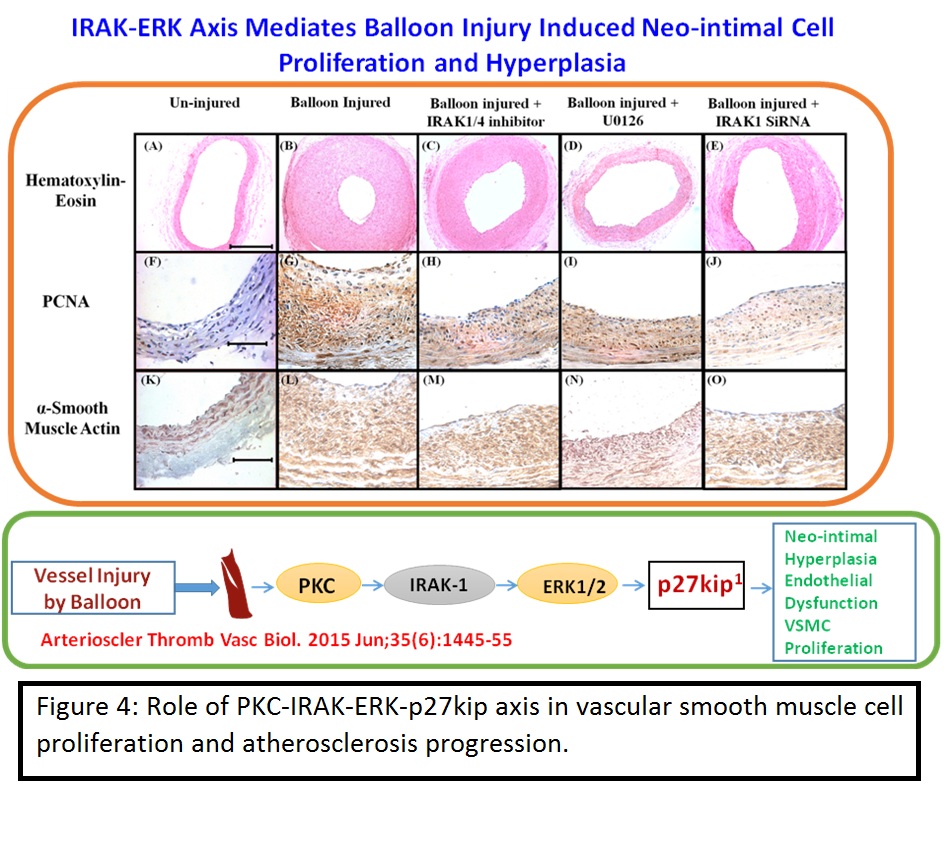
iii) Role of interleukin-1 receptor-associated kinase-1 in vascular smooth muscle cell proliferation and neointimal formation after rat carotid injury.
Having established that IRAK mediates Ox-LDL induced inflammation and macrophage foam cell formation, it was obvious to test its role in the progression of atherosclerotic cardiovascular disease in the animal models. Previous studies showed reduced frequency of atherosclerotic plaques in interleukin-1 receptor-associated kinase-1 (IRAK1)-deficient mice; however, the underlying mechanism was not clear. Therefore, this study investigated the role of IRAK1 in vascular smooth muscle cell proliferation and neointimal hyperplasia. Stimulation of rat primary vascular smooth muscle cells with fetal bovine serum (10%) or platelet-derived growth factor-BB (20 ng/mL) for 15 minutes to 24 hours induced a time-dependent increase in IRAK1 and extracellular signal-regulated kinase (ERK) activation, proliferating cell nuclear antigen upregulation and p27Kip1 downregulation as assessed by Western blotting. Inhibitors of ERK pathway (U0126, 10 μmol/L), IRAK (IRAK1/4, 3 μmol/L), protein kinase C (PKC; Ro-31-8220, 1 μmol/L), siRNA of toll-like receptor-4 (200 nmol/L), and PKC-ε (200 nmol/L) significantly attenuated these changes. Platelet-derived growth factor induced endogenous IRAK-ERK-PKC-ε association in a toll-like receptor-4 and PKC-ε-dependent manner. A time-dependent increase in IRAK1 and ERK activation was observed after 15 minutes, 30 minutes, 1 hour, 6 hours, 12 hours, and 24 hours of carotid balloon injury in rats. Balloon injury induced endogenous IRAK-ERK-PKC-ε interaction. Perivascular application of IRAK1/4 inhibitor (100 μmol/L), U0126 (100 μmol/L), and IRAK1 siRNA (220 and 360 nmol/L) in pluronic gel abrogated balloon injury-induced ERK phosphorylation, activation, and p27Kip1 downregulation. Hematoxylin and eosin staining and immunohistochemistry of proliferating cell nuclear antigen and smooth muscle actin demonstrated that balloon injury-induced intimal thickening and neointimal vascular smooth muscle cell proliferation were significantly abrogated in the presence of IRAK1/4 inhibitor, IRAK1 siRNA, and U0126.
Therefore it can be concluded that IRAK1 mediates vascular smooth muscle cell proliferation and neointimal hyperplasia by regulating PKC-ε-IRAK1-ERK axis (Arterioscler Thromb Vasc Biol. 2015 Jun;35(6):1445-55).
Therefore the above studies conclusively established the role of IRAK in monocytic inflammation, macrophage foam cell formation and atherosclerosis progression. The studies speculate IRAK as am attractive target for treating atherosclerotic cardiovascular diseases.
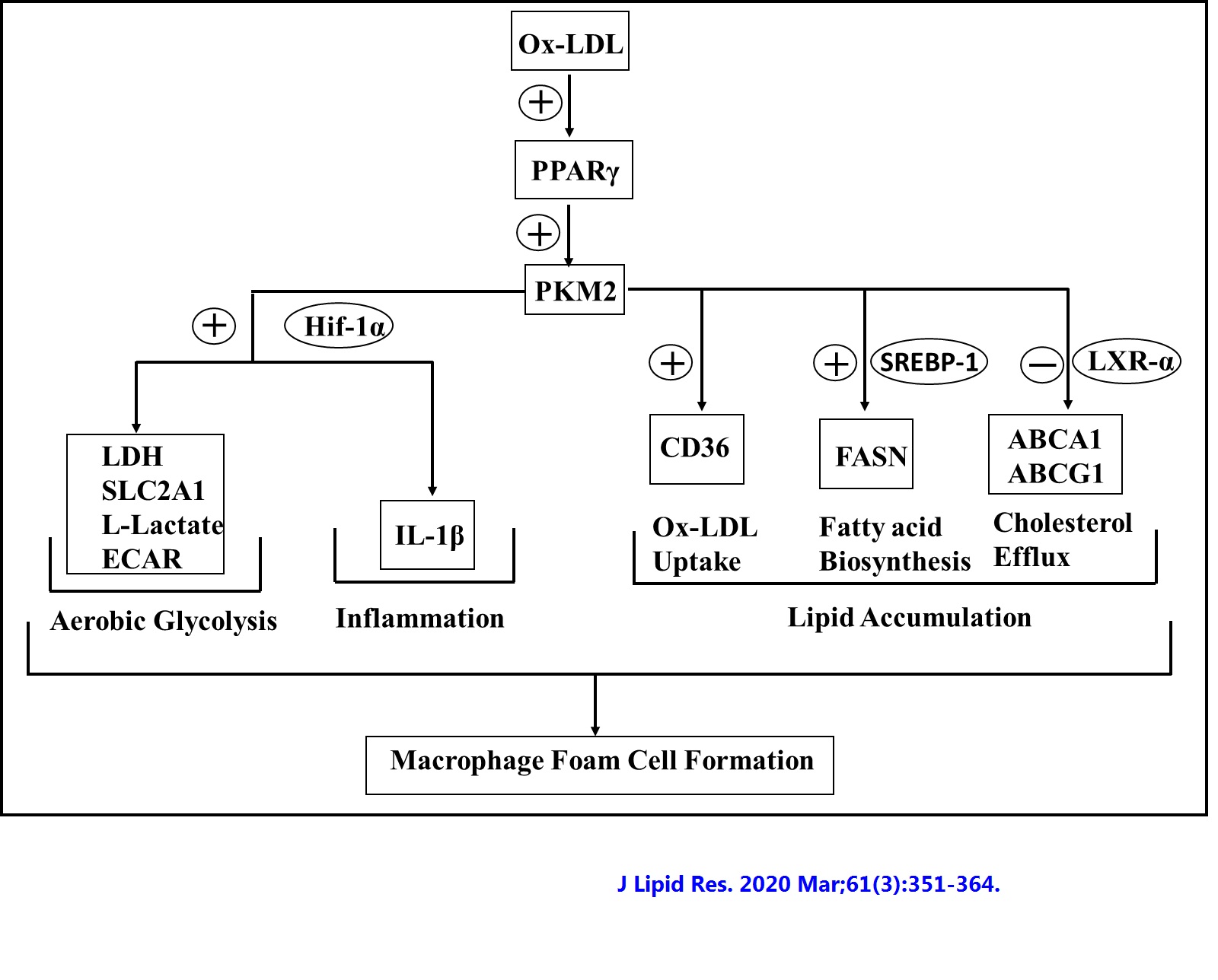
iv) Role of pyruvate kinase M2 in oxidized LDL-induced macrophage foam cell formation and inflammation
Pyruvate kinase M2 (PKM2) links metabolic and inflammatory dysfunction in atherosclerotic coronary artery disease; however, its role in oxidized LDL (Ox-LDL)-induced macrophage foam cell formation and inflammation is unknown and therefore was studied. In recombinant mouse granulocyte-macrophage colony-stimulating factor-differentiated murine bone marrow-derived macrophages, early (1-6 h) Ox-LDL treatment induced PKM2 tyrosine 105 phosphorylation and promotes its nuclear localization. PKM2 regulates aerobic glycolysis and inflammation because PKM2 shRNA or Shikonin abrogated Ox-LDL-induced hypoxia-inducible factor-1α target genes lactate dehydrogenase, glucose transporter member 1, interleukin 1β (IL-1β) mRNA expression, lactate, and secretory IL-1β production. PKM2 inhibition significantly increased Ox-LDL-induced ABCA1 and ABCG1 protein expression and NBD-cholesterol efflux to apoA1 and HDL. PKM2 shRNA significantly inhibited Ox-LDL-induced CD36, FASN protein expression, DiI-Ox-LDL binding and uptake, and cellular total cholesterol, free cholesterol, and cholesteryl ester content. Therefore, PKM2 regulates lipid uptake and efflux. DASA-58, a PKM2 activator, downregulated LXR-α, ABCA1, and ABCG1, and augmented FASN and CD36 protein expression. Peritoneal macrophages showed similar results. Ox-LDL induced PKM2- SREBP-1 interaction and FASN expression in a PKM2-dependent manner. Therefore, this study suggests a role for PKM2 in Ox-LDL-induced aerobic glycolysis, inflammation, and macrophage foam cell formation.
C) Role of inflammatory and metabolic pathways in Obesity and Heart Failure.
i) Cilostazol ameliorates heart failure with preserved ejection fraction (HFpEF) and diastolic dysfunction in obese and non-obese hypertensive mice.
Cilostazol (Ciloz) a potent Type III phosphodiesterase inhibitor is effective against inflammation, insulin resistance and cardiomyopathy. However, the effect of Ciloz on obesity-associated left ventricular diastolic dysfunction has not been explored yet. Hence, we examined the effect of Ciloz on cardiac remodelling and dysfunction in non-obese and obese-insulin resistant mice infused with AngiotensinII (AngII). Male C57BL/6 J mice were initially subjected to 19 weeks of chow or high fat diet (HFD) regimen and thereafter animals were randomised for AngII (1500 ng/kg/min, s.c) infusion or saline and Ciloz (50 mg/kg, p.o) for another 1 week.
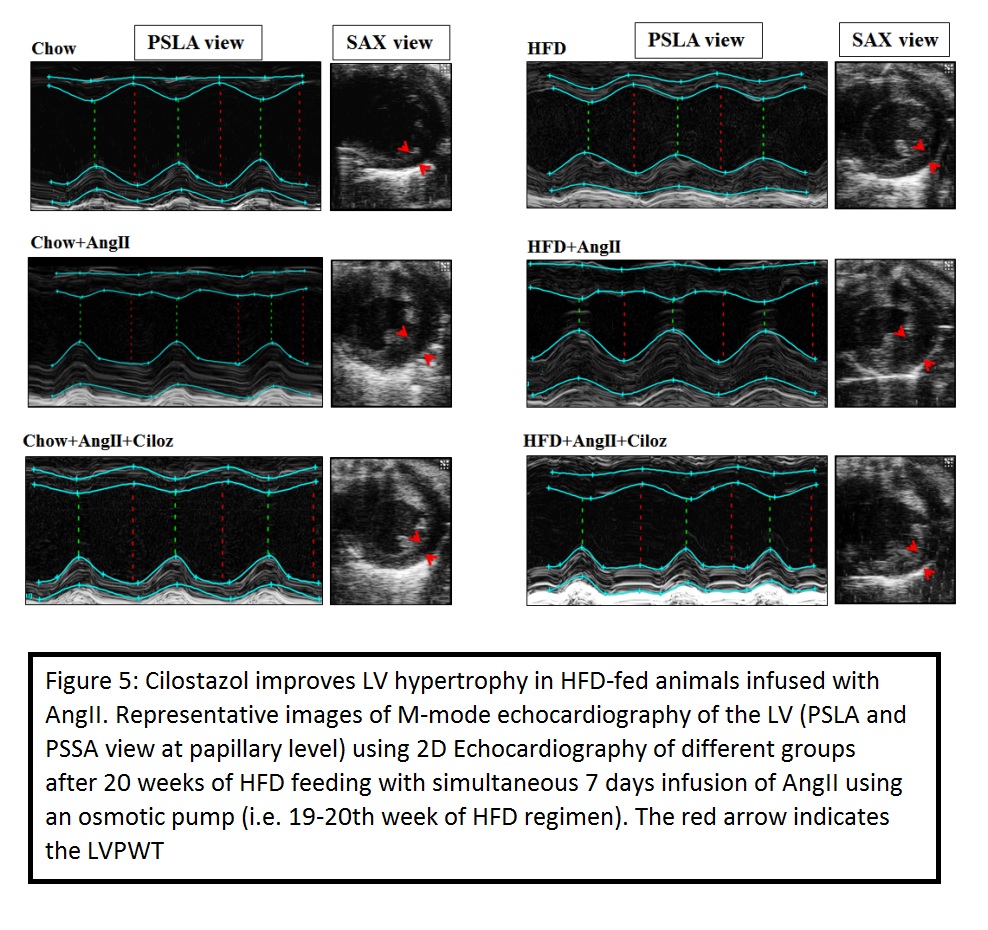
Obese and non-obese mice infused with AngII exhibited significant diastolic dysfunction and features of heart failure with preserved ejection fraction (HFpEF) since a decrease in fractional shortening and no change in ejection fraction were observed when compared with respective controls. Administration of AngII and Ciloz in HFD fed mice significantly improved the left ventricular function compared with AngII infused HFD mice as evinced from the echocardiographic data. Further, Ciloz treatment significantly reduced cardiomyocyte area, interstitial and perivascular fibrosis; and collagen deposition. Moreover, Ciloz reduced the inflammatory milieu in the heart as evinced by decreased F4/80+ and CD68+ cells; IL-1β and IL-6 gene transcripts. Quantitative assessment of the expression levels revealed substantial upregulation of MMP-9 (pro- and mature-forms) and α-SMA in the left ventricle of AngII infused HFD-fed mice, which was considerably suppressed by Ciloz regimen. The beneficial effect of Ciloz was associated with the normalization in gene expression of hypertrophic and fibrotic markers. Likewise, Ciloz administration markedly reduced the AngII and HFD induced TGF-β1/SMAD3 and Akt/mTOR signalling. Additionally, AngII administered and HFD-fed mice showed increased glycolytic flux, which was considerably diminished by Ciloz treatment as indicated from suppressed PKM2, HK-2, PDK-2, HIF-1α mRNA and GLUT-1 protein expression.
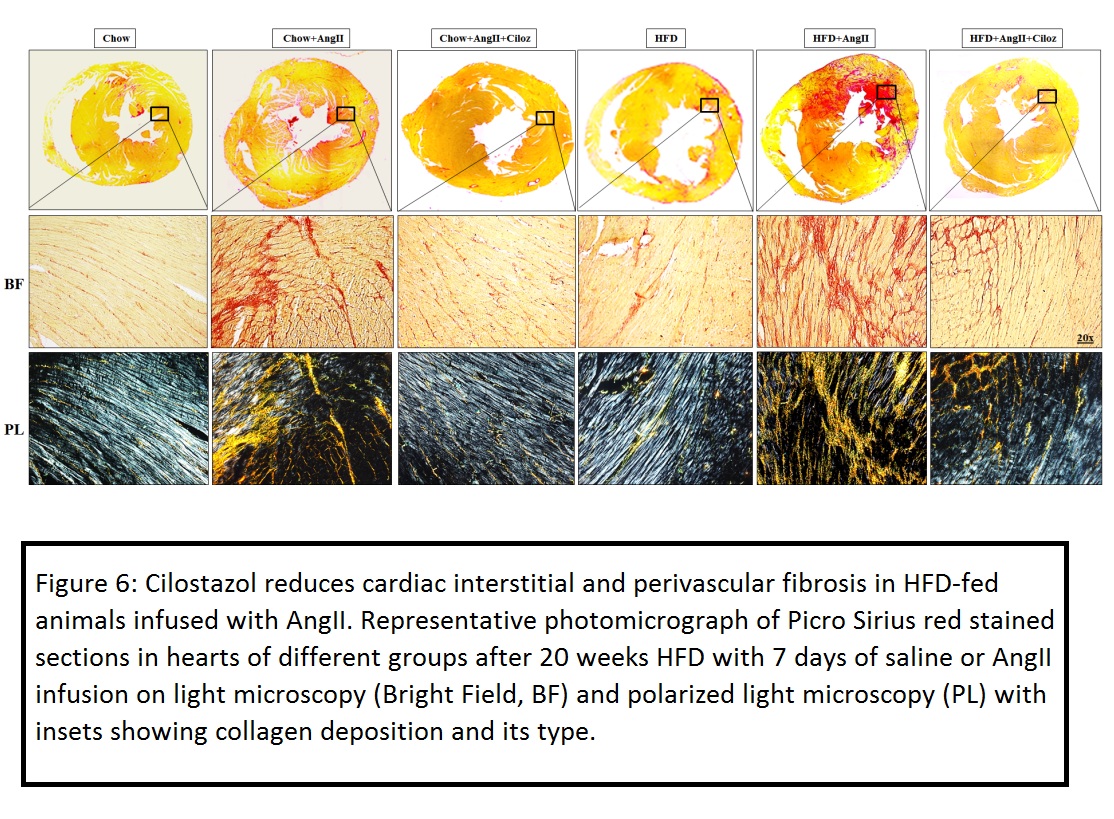
Taken together, Ciloz might be therapeutically exploited against AngII and obesity-associated diastolic dysfunction thereby preventing overt heart failure (J Mol Cell Cardiol. 2018 Aug 20).
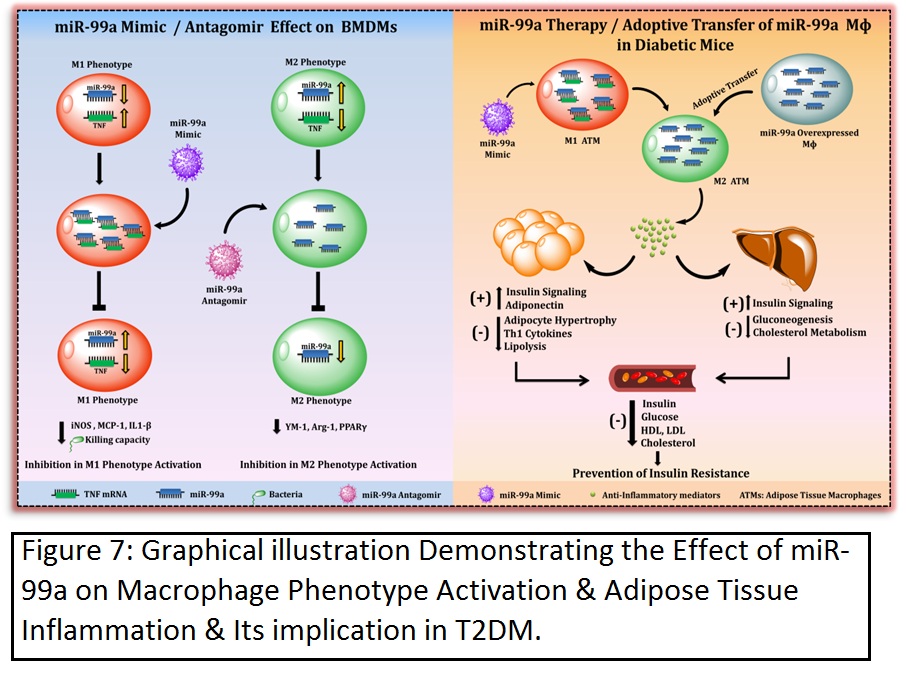
ii) MicroRNA-99a mimics inhibit M1 macrophage phenotype and adipose tissue inflammation by targeting TNFα.
In human adipose tissue and obesity, miR-99a expression is negatively correlated with inflammation. Therefore, the present study investigated the role of miR-99a in macrophage phenotype activation and adipose tissue inflammation. M2 BMDMs showed a significant increase in miR-99a expression when compared to the M0 and M1 phenotypes. Phenotype-switching experiments established an association between upregulated miR-99a expression and the M2 phenotype. Overexpression of miR-99a prevented M1 phenotype activation and attenuated bactericidal activity. Likewise, knockdown of miR-99a abolished M2 phenotype activation. By means of in silico target prediction tools and a luciferase reporter assay, TNFα was identified as a direct target of miR-99a. Knockdown of TNFα recapitulated the effect of miR-99a overexpression in M1 BMDMs. In a db/db mice model, miR-99a expression was reduced in eWAT and F4/80+ ATMs. Systemic overexpression of miR-99a in db/db mice attenuated adipocyte hypertrophy with increased CD301 and reduced CD86 immunostaining. Flow cytometry analysis also showed an increased M2 and a reduced M1 macrophage population. Mimics of miR-99a also improved the diabetic dyslipidemia and insulin signaling in eWAT and liver, with an attenuated expression of gluconeogenesis and cholesterol metabolism genes in the liver. Furthermore, adoptive transfer of miR-99a-overexpressing macrophages in the db/db mice recapitulated in vivo miR-99a mimic effects with increased M2 and reduced M1 macrophage populations and improved systemic glucose, insulin sensitivity, and insulin signaling in the eWAT and liver.
Therefore the present study demonstrates that miR-99a mimics can regulate macrophage M1 phenotype activation by targeting TNFα. miR-99a therapeutics in diabetic mice reduces the adipose tissue inflammation and improves insulin sensitivity (Cell Mol Immunol. 2018 May 30. doi: 10.1038/s41423-018-0038-7).
D) Novel herbal preparations for the treatment of atherosclerotic cardiovascular diseases.
i) Curcuma oil attenuates hyperlipidaemia, accelerated atherosclerosis and macrophage foam-cell formation by modulating genes involved in plaque stability, lipid homeostasis and inflammation.
In the present study, the anti-atherosclerotic effect and the underlying mechanism of curcuma oil (C. oil), a lipophilic fraction from turmeric (Curcuma longa L.), was evaluated in a hamster model of dyslipidaemia, accelerated atherosclerosis and in THP-1 macrophages. Male golden Syrian hamsters were fed a chow or high-cholesterol (HC) and fat-rich diet with or without C. oil (30, 100 and 300 mg/kg) for 28 d. In HC diet-fed hamsters, C. oil significantly reduced plasma total cholesterol, LDL-cholesterol and TAG, and increased HDL-cholesterol when compared with the HC group. Male golden Syrian hamsters were also subjected to partial carotid ligation (PCL) or FeCl3-induced arterial oxidative injury (Ox-injury) after 1 week of treatment with a high-cholesterol (HC) diet or HC diet plus C. oil (100 and 300 mg/kg, orally). Hamsters fed with the HC diet were analysed at 1, 3 and 5 weeks following carotid injury. The HC diet plus C. oil-fed group was analysed at 5 weeks. In hyperlipidaemic hamsters with PCL or Ox-injury, C. oil (300 mg/kg) reduced elevated plasma and aortic lipid levels, arterial macrophage accumulation, and stenosis when compared with those subjected to arterial injury alone. Similarly, elevated mRNA transcripts of matrix metalloproteinase-2 (MMP-2), MMP-9, cluster of differentiation 45 (CD45), TNF-α, interferon-γ (IFN-γ), IL-1β and IL-6 were reduced in atherosclerotic arteries, while those of transforming growth factor-β (TGF-β) and IL-10 were increased after the C. oil treatment (300 mg/kg). The treatment with C. oil prevented HC diet- and oxidised LDL (OxLDL)-induced lipid accumulation, decreased the mRNA expression of CD68 and CD36, and increased the mRNA expression of PPARα, LXRα, ABCA1 and ABCG1 in both hyperlipidaemic hamster-derived peritoneal and THP-1 macrophages. The administration of C. oil suppressed the mRNA expression of TNF-α, IL-1β, IL-6 and IFN-γ and increased the expression of TGF-β in peritoneal macrophages. In THP-1 macrophages, C. oil supplementation prevented OxLDL-induced production of TNF-α and IL-1β and increased the levels of TGF-β.
C. oil demonstrated an anti-hyperlipidaemic effect and reduced lipid-induced oxidative stress, platelet activation and vascular dysfunction. The anti-hyperlipidaemic effect exhibited by C. oil seems to be mediated by the modulation of PPARa, LXRa and associated genes involved in lipid metabolism and transport.
Overall the present study shows that C. oil attenuates Hyperlipidemia and arterial injury-induced accelerated atherosclerosis, inflammation and macrophage foam-cell formation (Br J Nutr. 2013 Aug 28;110(3):437-46, Br J Nutr. 2015 Jan 14;113(1):100-13).
References:
1) Tiwari, RL, Singh V and Barthwal MK Macrophages: An elusive yet emerging therapeutic target of atherosclerosis.. Medicinal Research Review. 2007 Nov 14; 28 (4):483-544
2) Barthwal MK. Macrophage Foam Cell Formation in Atherosclerosis: The Road Ahead, April 29, 2009. Invited commentary at official website of international atherosclerosis society - http://www.athero.org/.
3) Tiwari RL, Singh V, Singh A, Barthwal MK.IL-1R-associated kinase-1 mediates protein kinase Cδ-induced IL-1β production in monocytes. J Immunol. 2011 Sep 1;187(5):2632-45
4) Singh V, Jain M, Misra A, Khanna V, Rana M, Prakash P, Malasoni R, Dwivedi AK, Dikshit M, Barthwal MK. Curcuma oil ameliorates hyperlipidaemia and associated deleterious effects in golden Syrian hamsters. Br J Nutr. 2013 Aug 28;110(3):437-46
5) Tiwari RL, Singh V, Singh A, Rana M, Verma A, Kothari N, Kohli M, Bogra J, Dikshit M, Barthwal MK.PKCδ-IRAK1 axis regulates oxidized LDL-induced IL-1β production in monocytes. J Lipid Res. 2014 Jul;55(7):1226-44.
6) Jain M, Singh A, Singh V, Barthwal MK. Involvement of interleukin-1 receptor-associated kinase-1 in vascular smooth muscle cell proliferation and neointimal formation after rat carotid injury. Arterioscler Thromb Vasc Biol. 2015 Jun;35 (6):1445-55.
7) Singh V, Rana M, Jain M, Singh N, Naqvi A, Malasoni R, Dwivedi AK, Dikshit M, Barthwal MK.Curcuma oil attenuates accelerated atherosclerosis and macrophage foam-cell formation by modulating genes involved in plaque stability, lipid homeostasis and inflammation. Br J Nutr. 2015 Jan 14;113(1):100-13.
8) Singh A, Singh V, Tiwari RL, Chandra T, Kumar A, Dikshit M, Barthwal MK. The IRAK-ERK-p67phox-Nox-2 axis mediates TLR4, 2-induced ROS production for IL-1β transcription and processing in monocytes. Cell Mol Immunol. 2016 Nov;13(6):745-763. doi: 10.1038/cmi.2015.62. Epub 2015 Aug 31.
9) Rana M, Kumar A, Tiwari RL, Singh V, Chandra T, Dikshit M, Barthwal MK. IRAK regulates macrophage foam cell formation by modulating genes involved in cholesterol uptake and efflux. Bioessays. 2016 Jul;38(7):591-604.
10) Reddy SS, Agarwal H, Barthwal MK. Cilostazol ameliorates heart failure with preserved ejection fraction and diastolic dysfunction in obese and non-obese hypertensive mice. J Mol Cell Cardiol. 2018 Aug 20. pii: S0022-2828(18)30815-0. Doi.
11) Jaiswal A, Reddy SS, Maurya M, Maurya P, Barthwal MK.MicroRNA-99a mimics inhibit M1 macrophage phenotype and adipose tissue inflammation by targeting TNFα. Cell Mol Immunol. 2018 May 30. doi: 10.1038/s41423-018-0038-7.
12) Gupta P, Barthwal MK. IL-1 β genesis: the art of regulating the regulator. Cell Mol Immunol. 2018 Jun 19. doi: 10.1038/s41423-018-0054-7.